Overview
On April 12, 2018, the American Academy of Arts and Sciences presented the Francis Amory Prize to Barbara J. Meyer (University of California, Berkeley). An introduction by David C. Page (Whitehead Institute; Massachusetts Institute of Technology) and acceptance remarks from Barbara Meyer appear below.

Let me start by asking a question. Is anyone interested in sex? That may be too hard a question. How about is anyone interested in the differences between the sexes? I have had the pleasure of being a colleague of our Francis Amory Prize recipient, Barbara Meyer, since the early 1980s. Barbara and I – as well as her husband Tom Cline – share a passion for using the toolkits of genetics and molecular biology to understand the differences between the sexes.
I explore differences between males and females in our own species. This is sometimes a complicated matter. Barbara wisely explores differences between males, who are makers of sperm, and hermaphrodites, who are makers of both eggs and sperm. Barbara studies these differences in a very sexy species that modern biologists refer to simply as the worm. This is, of course, the nematode Caenorhabditis elegans. As the song goes, sex is a many splendored thing, and it presents scholars with an infinite array of nuances, distractions, and sideshows. Researchers are attracted to study sex in our own species, in the worm, and elsewhere.
Why has Barbara’s work had such impact? The answer is actually very simple: Barbara has never been distracted by the many sideshows. Instead, she has consistently pursued the most central, the most fundamental questions about males, females, and hermaphrodites. And she has framed her findings in a manner that resonates far beyond the community of what we, biologists, affectionately refer to as worm people.
The ability to frame the fundamental question and to connect the answer to all of life is what sets Barbara’s mind and her inquiry apart. And that is what we honor tonight.
It has been said that if we understand the worm, we understand life. Through your pioneering research using the amazing nematode worm C. elegans, you have shown the interplay between chromosome structure and function in sex determination. You have made groundbreaking discoveries with respect to how X chromosomes are counted to determine sexual fate, and how the related process of dosage compensation is achieved. And you have identified the master control gene involved in sex determination. Your research into the mechanisms underlying dosage compensation produced many key insights into gene regulation, influencing the work on all higher eukaryotes, including humans. And because of you, we truly have a better understanding of life.
You are a mentor to many. And your work has been praised for its elegant genetic analysis followed by beautiful molecular and cellular studies, which have continued to yield a deep and fascinating picture of these processes. You are a true inspiration to your colleagues and those who will follow.
As the citation for the Francis Amory Prize reads: “With relentless dedication, determination, and passion, you have delivered on science’s promise to provide a more expansive sense of who we are, how we fit with life on earth, and how we may improve the human condition.”

It is a great honor and pleasure to be amongst some of my best friends on the planet, and to be able to be in Cambridge, a place I love, to receive this honor and to share it with people I admire so much. I am really grateful for this opportunity to see you all.
Tonight’s lecture is a brief presentation about the fundamentals of sex and death. They are related in some interesting and in some boring ways. I will touch on both.
As David mentioned, we have taken our studies of sex and death all the way from the genetic underpinnings to in-depth molecular and biochemical studies. I will try to give you a flavor of the different approaches and conclusions.
The year 1921 was a landmark time when Calvin Bridges, a developmental geneticist, launched a series of experiments and publications on the topic of “Sex in Relation to Chromosomes and Genes.” He realized and became famous for understanding that a chromosomal signal determines sex in fruit flies. The idea of a chromosomal sex signal fascinated me. When I entered the worm field, I wanted to understand the basis of such sex signals.
Many mechanisms of sex determination exist. Although not all mechanisms are chromosomal, many are based on chromosome counting (Figure 1a). In humans, the Y chromosome, as David Page knows well, determines sex. In worms, as in fruit flies, sex is determined by the number of X chromosomes. These animals count sex chromosomes, and they count them against the background of other non-sex chromosomes called autosomes, which I will describe in a moment.
A virtue of studying sex determination is that it is an exquisitely sensitive developmental switch that is highly tractable when one thinks deeply about genetics and molecular mechanisms. The choice of sexual fate is one of the most fundamental, binary developmental decisions in biology, and one that most organisms, from bacteria to man, must make. The switch results in an obvious choice between two developmental fates: male/hermaphrodite in the case of the worm.
The worm is unusual in that it is adept at calculating fractions (Figure 1b). This worm not only understands how to count the number of X chromosomes, but it can count them with great accuracy and fidelity. You and I have two sets of non-sex chromosomes. We are diploid, and a worm in the wild is also diploid most of the time. A diploid worm that has one X chromosome is a male (1X to 2 sets of autosomes is an X:A ratio of 0.5). A diploid worm that has two X chromosomes is a hermaphrodite (2Xs to 2 sets of autosomes is an X:A ratio of 1). Impressively, this worm is more skillful than simply being able to count to two. It can calculate fractions. We can generate worms in the lab with unusual combinations of chromosomes, and remarkably, the worm can figure out the tiny differences in these ratios and commit, reproducibly, to becoming a fertile male or hermaphrodite. The worm translates ratios between 0.5 and 0.67 into the male fate, and ratios between 0.75 and 1 into the hermaphrodite fate.
That ability fascinated me. My desire to understand the mechanism launched my studies. What enabled us to figure out part of the answer is that we discovered the genetic and molecular target of this specific chromosome-counting mechanism (Figure 1c). We found a gene we named xol-1 for XO-lethal (male lethal). This is where the death part comes in. I will explain the cause of death shortly.
When an animal is programmed genetically to be a male, the level of xol-1 is high. When an animal is programmed genetically to be a hermaphrodite, the level of xol-1 is low. I wanted to understand what this switch did and how it knew what to do.
A pan balance shows the answer in simple form (Figure 1d). If a diploid organism (AA, two sets of autosomes) has two X chromosomes (XX), it becomes a hermaphrodite. If this diploid organism (AA) has only one X chromosome, it becomes a male. We were able to discover the features of the X chromosomes and autosomes that are counted. We found that specific genes encoded on X chromosomes, called X-signal elements (XSE), act as repressors of xol-1, and genes encoded on autosomes called autosomal-signal elements (ASE) act as activators of xol-1. The XSE repressor genes outcompete the ASE activator genes to repress xol-1 when present in two copies, due to two X chromosomes. Or they allow xol-1 to remain active when present in one copy. In that case, the ASE activators win. It is a simple switch. The complication is that the switch controls both sex and death.
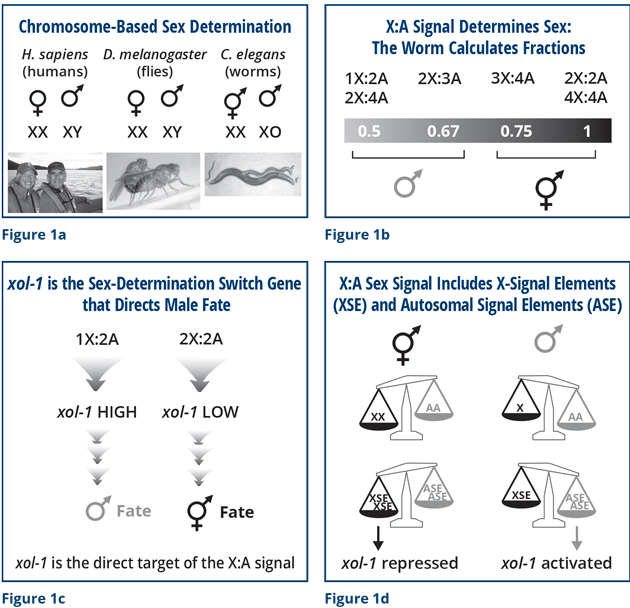
Although we discovered the molecular basis of the sex signal and its direct target, the master sex-determination-switch gene xol-1, a fundamental question remained: How is the signal interpreted reproducibly in an “all or none” manner to elicit either male or hermaphrodite development, never intersexual development? We have pioneered new approaches to answer these questions by developing methods with single-molecule and single-cell resolution. These methods enable us to count the number of molecules made from individual sex-determination genes – xol-1, XSE, and ASE – in every cell of an embryo and compare them across hundreds of embryos with different numbers of chromosomes.
The innovation that makes this analysis possible is a new network architecture for machine learning we are developing with James Sethian, a mathematics professor at the University of California, Berkeley. It automatically renders every cell in an embryo in 3 dimensions (3D) so that the total number of specifically labeled molecules in each cell can be counted. The network is trained by comparing a 3D stack of hundreds of high-resolution images of an embryo having all of its cell membranes lit up with a green fluorescent protein to a second 3D stack of training images made from the same embryo having all cell boundaries manually outlined to model intact membrane structure. The training set takes weeks of labor to prepare per embryo – an arduous task! Then, applying the newly trained neural network to a new 3D stack of images from a different embryo automatically segments the cell boundaries with stunning accuracy and speed (a few seconds). Once the cell boundaries are defined, quantification of all the labeled molecules in a cell can be automated.
Now we return to the connection between sex and death. This relationship is best understood in the context of some well-known human genetic disorders. I am sure you are all familiar with the condition called Down syndrome, in which three copies of chromosome 21 are present instead of two (Figure 2a). The elevated levels of gene products from the extra chromosome 21 cause numerous abnormalities, creating problems in development. Down syndrome is one of the few examples in which three copies of a human chromosome is even compatible with life. Three copies of almost any other chromosomes would cause a developing fetus to die.
In a similar vein, males and hermaphrodites in worm species require equivalent levels of X-chromosome products, despite the fact that males have only one X and hermaphrodites have two. As the nematode sex-determination mechanism evolved, a dosage compensation process co-evolved to equalize X expression between the sexes. We found that xol-1 controls not only sex determination, but also dosage compensation. If xol-1 is turned off accidentally in a male, he will die from an incorrect level of X-chromosome gene expression. Thus, sex and death are indeed linked.
Most organisms that determine sex by a chromosomal mechanism have co-evolved an X-chromosome dosage compensation process. The strategies differ (Figure 2b). For example, for every female in this room, a process called X-inactivation occurs. We have two X chromosomes, and one is inactivated randomly in every cell throughout our lifetimes. If X inactivation fails to occur in the fetus, she will die. In the fruit fly, the dosage compensation mechanism turns up expression from the single male X. Failure to turn up expression of the male X will cause the male to die. In the worm, we showed that a dosage compensation mechanism turns down expression of both hermaphrodite X chromosomes by half. A hermaphrodite that fails to turn on dosage compensation will arrest development as an embryo and die. Understanding how the worm dosage compensation process occurs is a daunting problem to solve, and we have been able to tackle it through the combination of multiple different approaches.
Genetically, we learned that when xol-1 is activated in a male, it turns off a hermaphrodite-specific gene called sdc-2 (sex determination and dosage compensation) (Figure 2c). In so doing, it turns on the pathway of male sexual differentiation and prevents the dosage compensation process from turning down expression of the single male X. When xol-1 is turned off in the hermaphrodite by the XSEs, sdc-2 is active; sdc-2 then turns on the pathway of hermaphrodite sexual differentiation and triggers the process of dosage compensation to turn down gene expression from both hermaphrodite X chromosomes.
Through genetics and biochemistry, we discovered a huge complex of proteins (including SDC-2) that binds to both hermaphrodite X and turns down expression of genes by half (Figure 2d). The worm cleverly co-opted and repurposed several proteins used in other essential biological processes from yeast to man. The co-opted proteins were originally required to compact and resolve replicated chromosomes in preparation for their segregation into daughter cells during somatic cell division (mitosis) or into sperm or eggs to make fertile gametes (meiosis). The worm co-opted the proteins for the entirely new purpose of regulating gene expression. We were able to find all the genes encoding these proteins by powerful genetic screens in which we searched for mutations that prevented xol-1 mutant males from dying. Not only did the worm steal many proteins for a new role, it continued to use these proteins in their old roles of compacting chromosomes in preparation for their segregation. The worm co-opted the ancient proteins for new roles via the evolution of worm-specific proteins that associate with the conserved chromosome compaction proteins and recruit them to X chromosomes at the appropriate time in embryogenesis, giving them two roles in the same cell (Figure 2d).
We showed that these promiscuous dosage compensation complex (DCC) proteins are recruited specifically to hermaphrodite X chromosomes by the XX-specific protein SDC-2, which triggers binding to cis-acting regulatory elements on X called rex sites (recruitment elements on X) to reduce gene expression by half (Figures 2e and f). The strongest of these sites have DNA sequence motifs that are highly enriched on X and confer specificity for SDC-2 binding to X. The complex then spreads along the entire X chromosome to sites that lack the X-enriched sequences (Figure 2f). XOL-1 prevents the DCC from binding to the single X chromosome of males by turning off SDC-2. If XOL-1 is defective, SDC-2 recruits the DCC to the male X, reduces gene expression, and kills all males, showing the connection between sex and death.
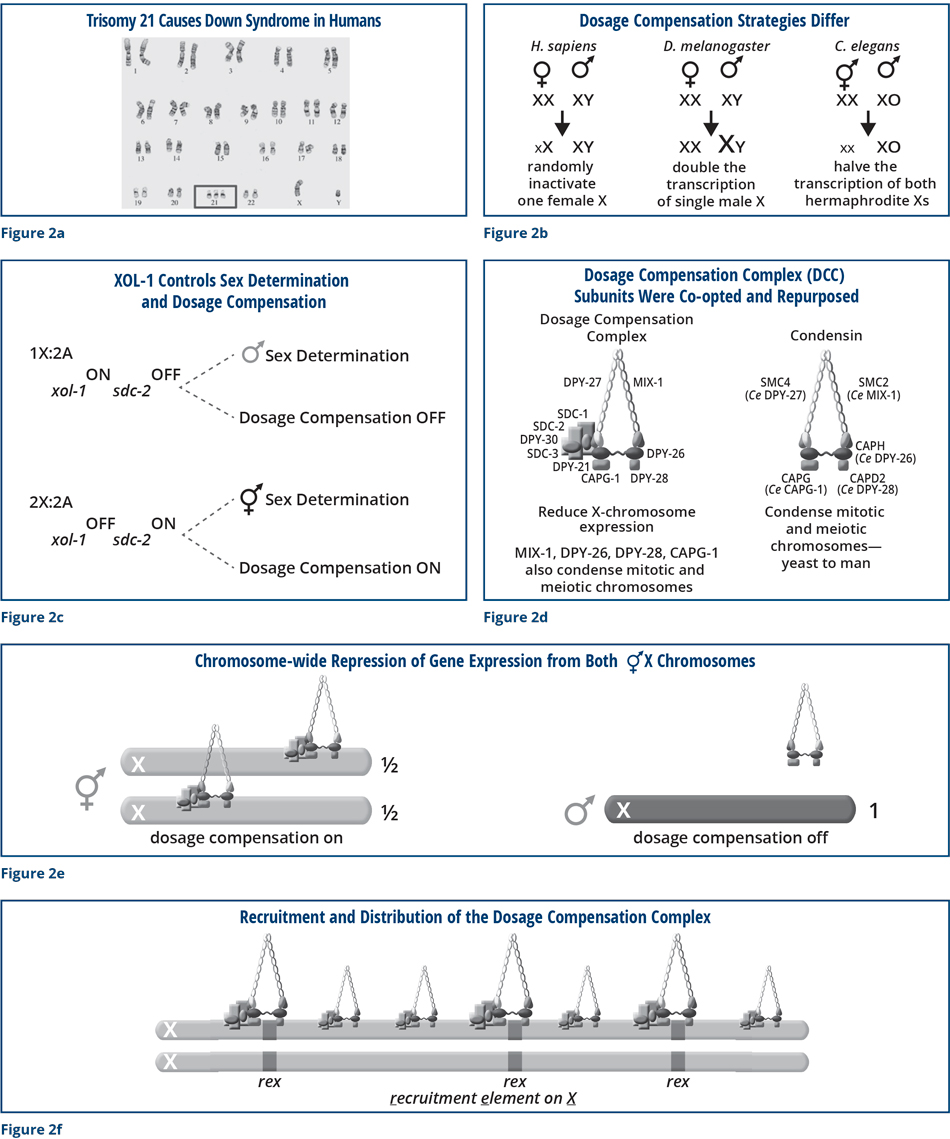
To help conceptualize the molecular function of the dosage compensation complex, the individual images in Figure 3 compare chromosome structure and segregation in wild-type embryos and embryos that are defective in one of the ancient chromosome segregation proteins from which the DCC was derived. (HCP-6 is related to DPY-28.) A green fluorescent histone protein that binds along the lengths of all chromosomes was used to visualize the chromosomes. The first images show a one-cell embryo just after a sperm (s) fertilized the oocyte (o), and the oocyte chromosome number was reduced to one copy (Figure 3a) by segregation of the extra copies into discarded structures called polar bodies (p). In the wild-type embryo, individual chromosomes condensed into distinct rod-shaped bodies as the sperm and oocyte chromosomes migrated toward each other (Figure 6b). In mutants, individual chromosomes remained disorganized and stringy during migration, but eventually achieved compaction somewhat similar to wild-type chromosomes as they aligned before for their segregation into daughter cells (Figure 3c). Chromosomes from wild-type embryos separated suddenly and completely during this first cell division, but mutant chromosomes failed to resolve their connections and remained attached by chromosome bridges (Figures 3d and e). Ultimately, segregated wild-type chromosomes partitioned equally into the two daughter cells and then decondensed. In contrast, mutant chromosomes never fully separated but instead remained strongly interconnected such that the two daughter cells received different chromosomal contents, neither of which had the correct number or type of chromosomes (Figure 3f). Mutant embryos eventually died from improper chromosome content due to chromosome segregation problems. Clearly, these ancient proteins are essential for proper chromosome compaction, resolution, segregation, and thus embryo viability.
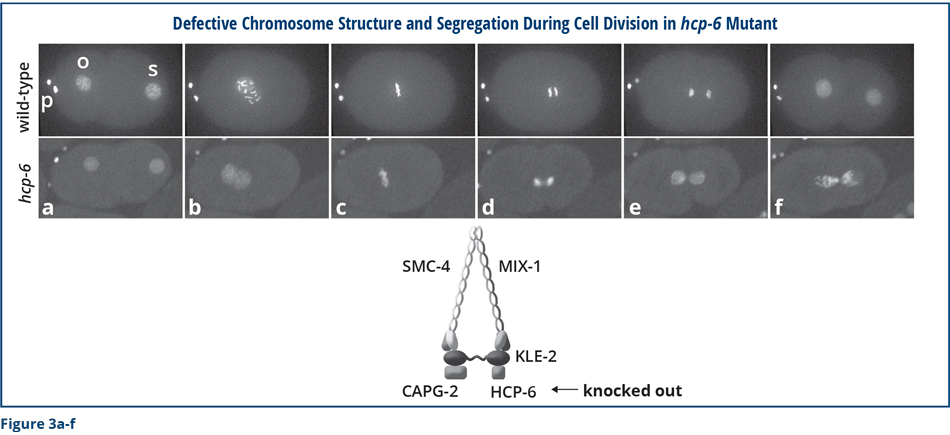
Both the similarity between DCC proteins and ancient chromosome segregation proteins, and the participation of DCC proteins in chromosome compaction, resolution, and segregation suggested that the DCC might influence X-chromosome gene expression by regulating the structure of X chromosomes.
We performed a series of experiments to determine whether the DCC reshapes the structure of X chromosomes and found that it does (Figure 4a). The DCC remodels hermaphrodite X chromosomes into a unique, sex-specific spatial conformation, distinct from that of autosomes or male X chromosomes, by using its strongest binding sites (rex sites) to facilitate long-range interactions among regions of DNA across X. The dosage-compensated X chromosomes have self-interacting domains called topologically associating domains (TADs) (Figure 4a). Sites on the DNA within each domain can interact, but a region of DNA in one domain cannot interact with a region in the other. In mammals, this type of structure restricts the genes and regulatory DNA that can associate with each other. In so doing, the structure facilitates proper development of an animal’s body plan and prevents inappropriate gene activation that causes defective development.
We found that most of the TAD boundaries on X coincide with a strong rex site (Figure 4a) and that disrupting the SDC-2 component of the DCC prevented the structure from forming. Importantly, we were able to use the modern techniques of genome editing to delete a rex site at a boundary and discovered that the TAD boundary was lost completely (Figure 4b). Thus, the rex site is essential for the formation of the boundary. Moreover, when we simultaneously deleted all of the rex sites at the DCC-dependent TAD boundaries (eight in total), the unique structure of X was destroyed. The X changed from a highly ordered structure into a more random structure. I have drawn these results as a cartoon, but you can trust me that we have large data sets that underlie this conclusion!
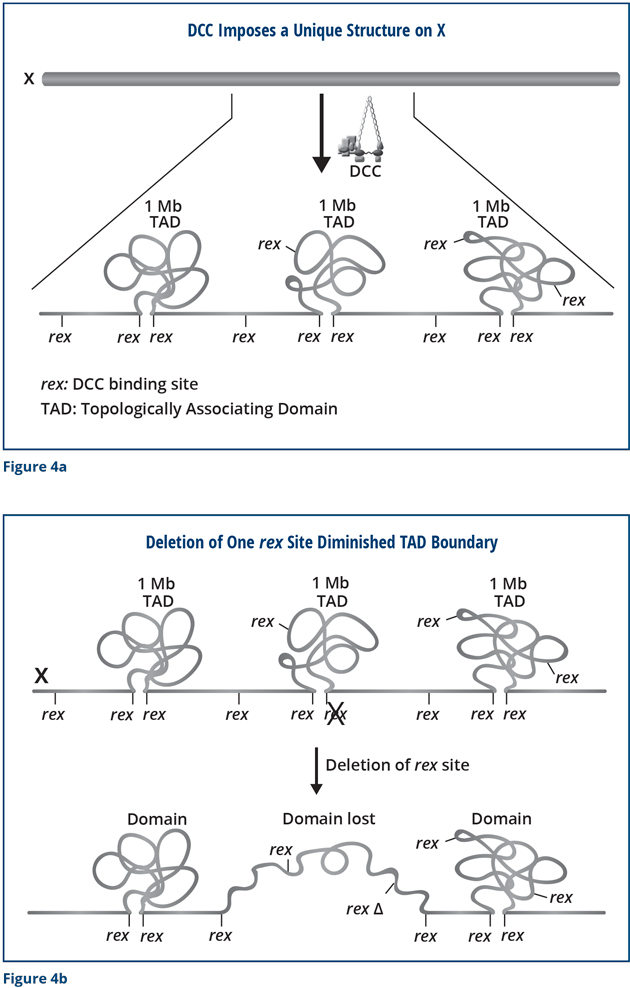
We were surprised to find that the hermaphrodites were still alive, even though the structure of their X chromosomes was disrupted. However, further analysis revealed that the lifespan of the mutant worms was not normal: the mutant worms died prematurely. They had progeria. Their lifespans were reduced by 20 percent. Moreover, the mutant worms were more sensitive to heat and other stresses than wild-type worms. The reduced tolerance to heat caused 40 percent more of the mutant animals to die than wild-type animals. Thus, the special structure of X seems to be important to enable the worm to have a normal life. Without the structure, the worms undergo premature death and are less tolerant to heat and other stresses.
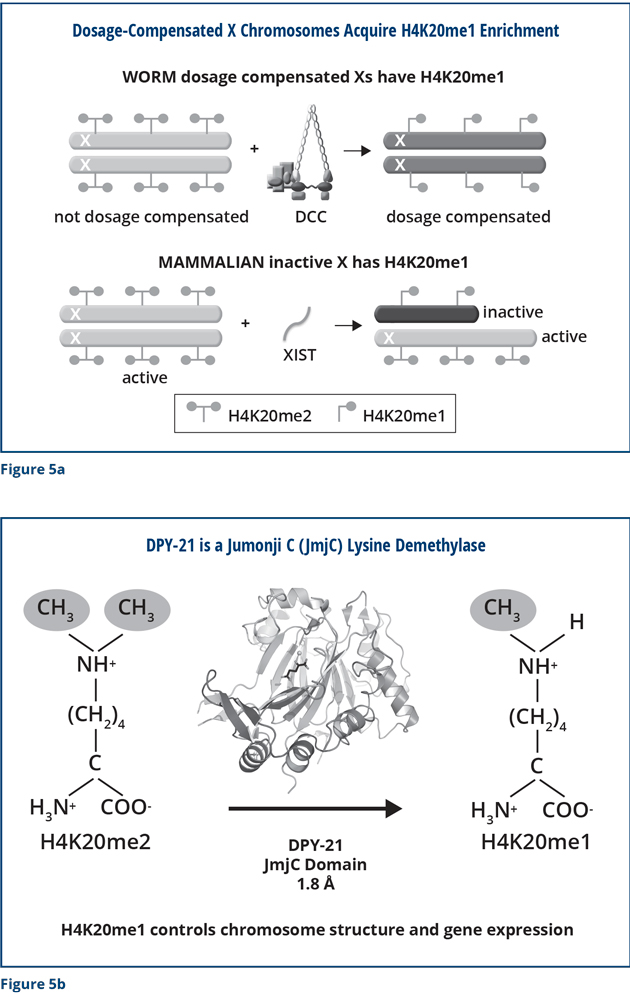
We also found that the DCC modifies X chromosomes in other important ways that are critical for proper X expression. We discovered that the DCC modifies the chemical composition of histone proteins on the X chromosome (Figure 5a). Dosage-compensated X chromosomes have a unique signature: the amino acid lysine 20 in the histone H4 protein carries one methyl group. The male X chromosome and dosage-compensation-defective hermaphrodite X chromosomes are different. Their lysine 20 in histone H4 carries two methyl groups. We discovered through X-ray crystallography, biochemistry, and imaging that the DPY-21 subunit of the DCC has an enzymatic activity that specifically removes one of the methyl groups on lysine 20 of histone H4. DPY-21 has a Jumonji C lysine demethylase activity that converts H4K20me2 to H4K20me1 (Figure 5b). Using genome engineering to change a single amino acid in DPY-21 that is important for this catalytic activity eliminated the enrichment of H4K20me1 on X. It also caused elevated X-chromosome gene expression, a hallmark of defective dosage compensation, reduced X-chromosome compaction, and disrupted X-chromosome conformation by diminishing the formation of TADs.
These studies are directly related to the development of mammals. As I mentioned previously, human females have one active X chromosome and one inactive X. The inactive mammalian X has the same chemical modification as the dosage-compensated worm X chromosomes. The inactive X has H4K20me1, but the active one has H4K20me2 (Figure 5a). We discovered that mammals have a protein that is very similar to DPY-21 and demonstrated in vitro that the mammalian protein has the same enzymatic activity as DPY-21. It converts H4K20me2 to H4K20me1, just as in worms, making it likely that the mammalian protein influences the composition and activity of the inactive X. Our findings emphasize the important concept that for many aspects of biology, understanding a process in humans relies on lessons learned via complementary studies in model organisms.
Analysis of nematode sex is also invaluable for understanding other aspects of biology, particularly evolution. My final remarks will then be about evolution. Thus far I have discussed how chromosome content dictates sexual fate and X-chromosome gene expression. But let me now describe surprising results about the converse: The mode of sexual reproduction (hermaphrodite vs. male/female) dictates genome size and content. This work, in collaboration with Eric Haag’s lab (University of Maryland), Asher Cutter’s lab (University of Toronto), and Erich Schwarz (Cornell University), was featured recently in The New York Times Science Trilobites section (Figure 6a). In the nematode genus Caenorhabditis, most species are male/female, but self-fertile hermaphroditic species like C. elegans have arisen independently at least three times. These hermaphroditic species are able to produce males but rarely do so in the wild. Uniparental reproduction via self-fertilizing hermaphrodites is thought to have been more adaptive than biparental reproduction via mating for survival in isolated habitats in which males and food were scarce, at least over the relatively short term.
We found that loss of virile males in the species was accompanied by the rapid shrinkage of the genome and loss of genes biased for expression in males (Figure 6b). At least 25 percent of the genome was lost from hermaphrodite species compared to closely related male/female species. Among the genes lost were those that encoded the family of MSS (Male Secreted Short) sperm proteins. They are glycoproteins found on the surface of male sperm. None of the hermaphrodite species has these genes, although small, inactive remnants of the genes were found. All the male/female species have these genes. Why are these genes important?

The MSS sperm-specific proteins are essential for enabling sperm to compete with other sperm, win the fight, and fertilize the oocyte. Remarkably, removing this mss gene family from the male/female species C. remanei by genome editing rendered the mutant male sperm unable to compete with normal sperm from the wild-type species, even though the mutant sperm could successfully fertilize oocytes in the absence of the normal sperm. A series of competitive mating experiments showed this phenomenon. If the wild-type male was allowed to mate first and the mutant male second, the wild-type male sired the progeny. Even if the mutant male mated first and the wild-type male mated second, the wild-type male sired the progeny. If the mutant male was the only male allowed to mate, he sired a normal number of progeny.
An even more compelling experiment was to take the mss gene family from the male/female species C. nigoni and add it to its closely related hermaphrodite species C. briggsae (diverged by only about one million years) and ask if the MSS-containing sperm became more potent and thus more competitive. The MSS-containing sperm always won the competition! C. briggsae males with the MSS sperm protein outcompeted C. briggsae males lacking the MSS sperm protein no matter if the MSS-containing males mated first or second. Finally, a mating between wild-type C. briggsae males and hermaphrodites yielded 50 percent cross-progeny males in the first generation, as expected. If their progeny was allowed to propagate continuously until the twelfth generation, no males persisted in the population: the hermaphrodite mode of replication won out. In contrast, while a mating between mss mutant C. briggsae males and hermaphrodites also yielded 50 percent cross-progeny males in the first generation, by the twelfth generation, 30 percent of the offspring was still male, demonstrating a marked increase in the effectiveness of these males versus hermaphrodites.
These experiments demonstrate that the MSS protein family is key for sperm fitness and competitiveness, and hence male virility. Why would a species give up males? What is the advantage of being a self-fertile hermaphrodite species? In isolated environments where food is limited and males are scarce, reproduction and survival are more successful if all progeny can be produced by a single parent. To reach that state, genes important for efficient reproduction through mating must be eliminated. The dramatic transition in mode of reproduction is a compelling evolutionary adaptation for survival.
I’ll end with that thought. I thank my lab members and funding sources throughout the decades for conducting and enabling this exciting research. Thank you for listening and thank you for this award.
© 2018 by David C. Page and Barbara J. Meyer, respectively
To view or listen to the presentations, visit www.amacad.org/events/annual-awards-ceremony-2018